
Identification and characterization of co-variations in VHL and TP53 in a family with Von Hippel-Lindau disease
-
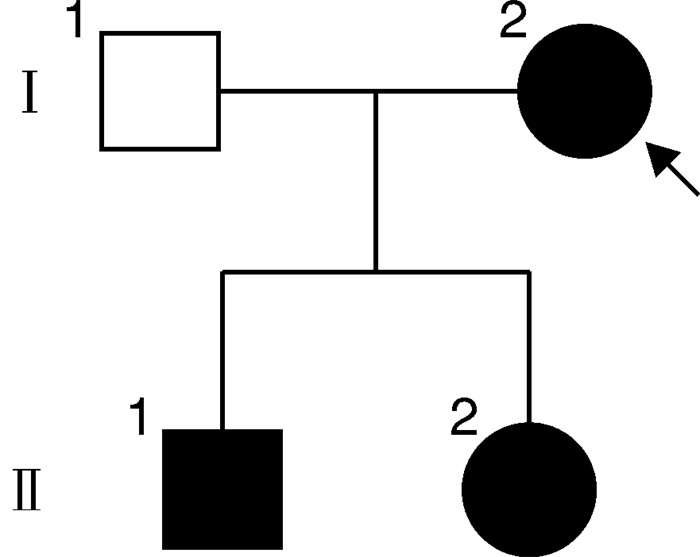
摘要: 目的 研究Von Hippel-Lindau(VHL)综合征合并VHL基因和TP53 基因突变家系的临床表型和基因突变分析。方法 回顾分析患有VHL综合征的家系,收集临床资料,对该家系进行全外显子组测序(WES)和Sanger测序验证,同时进行生物信息学分析。对肾上腺肿块进行苏木精-伊红(HE)染色观察结构改变。结果 先证者无典型嗜铬细胞瘤临床表现,肾上腺素和去甲肾上腺素均在正常范围,CT示双侧肾上腺肿块和膀胱肿块。先证者的儿子阵发性出汗病史伴有去甲肾上腺素升高,CT示双侧肾上腺肿块。先证者右侧肾上腺肿块病理检查示血管瘤伴局部嗜铬细胞增生,左侧肾上腺肿块示嗜铬细胞瘤。先证者儿子双侧肾上腺病理检查示嗜铬细胞瘤。基因检测和分析提示VHLc.491A>G(p.Gln164Arg)和TP53 c.91G>A(p.Val31Ile)共突变,分别被认为是致病性和不确定意义。结论 通过基因检测发现患有VHL综合征家系合并VHL和TP53 突变,有助于扩展对VHL和TP53 基因突变的认识,未来需要进一步研究以更好地理解VHL和TP53 基因共突变的协同效应。Abstract: Objective To study a family with Von Hippel-Lindau(VHL) disease with a rare co-variation in VHL and TP53 .Methods The clinical data were collected. The Whole-exome sequencing(WES) and Sanger sequencing were conducted in this family and bioinformatics analysis were performed. Variations were interpreted and hematoxylin and eosin(H&E) staining of adrenal mass was carried out to evaluate the affected cases.Results Bilateral adrenal mass and a bladder mass were detected in the proband without typical clinical manifestation of pheochromocytoma. Bilateral adrenal masses were detected in the son of the proband with elevated norepinephrine and paroxysmal sweating. Genetic analysis revealed a rare co-variations in VHL c.491A>G(p.Gln164Arg) and TP53 c.91G>A(p.Val31Ile) which were deemed pathogenic and uncertain significance, respectively.Conclusion We identified a rare co-variation of VHL and TP53 in a family with VHL disease, presenting a distinct biomedical and clinical phenotype. Therefore, our study contributes to expanding the knowledge of variations in VHL and TP53 as well as providing new insights into the etiology of VHL disease. Further research is needed to better understand the synergistic effect of co-variations of VHL and TP53 .
-
Key words:
- Von Hippel-Lindau disease /
- TP53 /
- clinical phenotype /
- geneticvariability
-

-
表 1 患者临床特征及实验室检查结果
项目 Ⅰ2 Ⅱ1 参考值 性别 女 男 年龄/岁 40 17 血压/mmHg 120/83 105/60 心率/(次/min) 88 84 血钾/(mmol/L) 3.90 4.50 3.50~5.30 ACTH(9 am)/(pg/mL) 32.60 32.90 6.00~40.00 皮质醇(9 am)/(nmol/L) 247.44 928.20 145.40~619.40 皮质醇(4 pm)/(nmol/L) 123.06 184.00 94.90~462.40 醛固酮/(pg/mL) 69.70 101.00 70.00~300.00 血管紧张素Ⅰ/(ng/mL) 1.67 4.46 0.10~6.56 血管紧张素Ⅱ/(pg/mL) 96.60 70.90 50.00~120.00 PRA/(ng/mL/h) 1.13 16.64 0.10~6.56 24 h尿去甲肾上腺素/(μg/24 h) 20.66 2 120.48 10.00~80.00 24 h尿多巴胺/(μg/24 h) 54.12 857.39 53.00~493.00 24 h尿肾上腺素/(μg/24 h) 16.07 23.41 < 20.00 去甲肾上腺素/(ng/L) 855.62 1 987.34 0~1 700.00 多巴胺/(ng/L) 116.32 97.96 0~200.00 肾上腺素/(ng/L) 180.71 131.57 0~280.00 NMN/(nmol/L) 6.01 12.53 ≤0.89.00 24 h尿17-羟皮质醇/(mg/24 h) 5.87 8.50 2.00~10.00 24 h尿17-酮类固醇/(μmol/24 h) 14.41 13.73 6.00~25.00 第1次手术后 皮质醇(9 am)/(nmol/L) 344.57 315.48 145.40~619.40 ACTH(9 am)/(pg/mL) 6.00~40.00 第2次手术后 皮质醇(9 am)/(nmol/L) 63.28 94.61 145.40~619.40 ACTH(9 am)/(pg/mL) 529.50 6.00~40.00 注:ACTH:促肾上腺皮质激素; PRA:血浆肾素活性; NMN:甲氧基去甲肾上腺素。  下载: 导出CSV
下载: 导出CSV
-
[1] Kaelin WG. Von Hippel-Lindau disease[J]. Annu Rev Pathol, 2007, 2: 145-173. doi: 10.1146/annurev.pathol.2.010506.092049
[2] Maher ER, Iselius L, Yates JR, et al. Von Hippel-Lindau disease: a genetic study[J]. J Med Genet, 1991, 28(7): 443-447. doi: 10.1136/jmg.28.7.443
[3] Maher ER, Yates JR, Harries R, et al. Clinical features and natural history of von Hippel-Lindau disease[J]. Q J Med, 1990, 77(283): 1151-1163.
[4] Crossey PA, Richards FM, Foster K, et al. Identification of intragenic mutations in the von Hippel-Lindau disease tumour suppressor gene and correlation with disease phenotype[J]. Hum Mol Genet, 1994, 3(8): 1303-1308. doi: 10.1093/hmg/3.8.1303
[5] Kern SE, Kinzler KW, Bruskin A, et al. Identification of p53 as a sequence-specific DNA-binding protein[J]. Science, 1991, 252(5013): 1708-1711. doi: 10.1126/science.2047879
[6] Surget S, Khoury MP, Bourdon JC. Uncovering the role of p53 splice variants in human malignancy: a clinical perspective[J]. Onco Targets Ther, 2013, 7: 57-68.
[7] Park KJ, Choi HJ, Suh SP, et al. Germline TP53 Mutation and Clinical Characteristics of Korean Patients With Li-Fraumeni Syndrome[J]. Ann Lab Med, 2016, 36(5): 463-468. doi: 10.3343/alm.2016.36.5.463
[8] Yamada H, Shinmura K, Okudela K, et al. Identification and characterization of a novel germ line p53 mutation in familial gastric cancer in the Japanese population[J]. Carcinogenesis, 2007, 28(9): 2013-2018. doi: 10.1093/carcin/bgm175
[9] Niparuck P, Police P, Noikongdee P, et al. TP53 mutation in newly diagnosed acute myeloid leukemia and myelodysplastic syndrome[J]. Diagn Pathol, 2021, 16(1): 100. doi: 10.1186/s13000-021-01162-8
[10] Hou HA, Chou WC, Kuo YY, et al. TP53 mutations in de novo acute myeloid leukemia patients: longitudinal follow-ups show the mutation is stable during disease evolution[J]. Blood Cancer J, 2015, 5(7): e331. doi: 10.1038/bcj.2015.59
[11] Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology[J]. Genet Med, 2015, 17(5): 405-424. doi: 10.1038/gim.2015.30
[12] Sovinz P, Urban C, Uhrig S, et al. Pheochromocytoma in a 2.75-year-old-girl with a germline von Hippel-Lindau mutation Q164R[J]. Am J Med Genet A, 2010, 152A(7): 1752-1755. doi: 10.1002/ajmg.a.33407
[13] Ong KR, Woodward ER, Killick P, et al. Genotype-phenotype correlations in von Hippel-Lindau disease[J]. Hum Mutat, 2007, 28(2): 143-149. doi: 10.1002/humu.20385
[14] Eng C, Crossey PA, Mulligan LM, et al. Mutations in the RET proto-oncogene and the von Hippel-Lindau disease tumour suppressor gene in sporadic and syndromic phaeochromocytomas[J]. J Med Genet, 1995, 32(12): 934-937. doi: 10.1136/jmg.32.12.934
[15] Chen F, Kishida T, Yao M, et al. Germline mutations in the von Hippel-Lindau disease tumor suppressor gene: correlations with phenotype[J]. Hum Mutat, 1995, 5(1): 66-75. doi: 10.1002/humu.1380050109
[16] Landrum MJ, Lee JM, Riley GR, et al. ClinVar: public archive of relationships among sequence variation and human phenotype[J]. Nucleic Acids Res, 2014, 42(Database issue): D980-D985.
[17] Ricker CA, Crawford K, Matlock K, et al. Defining an embryonal rhabdomyosarcoma endotype[J]. Cold Spring Harb Mol Case Stud, 2020, 6(2): a005066. doi: 10.1101/mcs.a005066
[18] Gruber LM, Erickson D, Babovic-Vuksanovic D, et al. Pheochromocytoma and paraganglioma in patients with neurofibromatosis type 1[J]. Clin Endocrinol(Oxf), 2017, 86(1): 141-149. doi: 10.1111/cen.13163
[19] Dluhy RG. Pheochromocytoma--death of an axiom[J]. N Engl J Med, 2002, 346(19): 1486-1488. doi: 10.1056/NEJM200205093461911
[20] Eisenhofer G, Walther MM, Huynh TT, et al. Pheochromocytomas in von Hippel-Lindau syndrome and multiple endocrine neoplasia type 2 display distinct biochemical and clinical phenotypes[J]. J Clin Endocrinol Metab, 2001, 86(5): 1999-2008. doi: 10.1210/jcem.86.5.7496
[21] Walther MM, Reiter R, Keiser HR, et al. Clinical and genetic characterization of pheochromocytoma in von Hippel-Lindau families: comparison with sporadic pheochromocytoma gives insight into natural history of pheochromocytoma[J]. J Urol, 1999, 162(3 Pt 1): 659-664.
[22] Li SR, Nicholson KJ, Mccoy KL, et al. Clinical and Biochemical Features of Pheochromocytoma Characteristic of Von Hippel-Lindau Syndrome[J]. World J Surg, 2020, 44(2): 570-577. doi: 10.1007/s00268-019-05299-y
[23] Maxwell PH, Wiesener MS, Chang GW, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis[J]. Nature, 1999, 399(6733): 271-275. doi: 10.1038/20459
[24] Clifford SC, Cockman ME, Smallwood AC, et al. Contrasting effects on HIF-1alpha regulation by disease-causing pVHL mutations correlate with patterns of tumourigenesis in von Hippel-Lindau disease[J]. Hum Mol Genet, 2001, 10(10): 1029-1038. doi: 10.1093/hmg/10.10.1029
[25] Stebbins CE, Kaelin WG Jr, Pavletich NP. Structure of the VHL-ElonginC-ElonginB complex: implications for VHL tumor suppressor function[J]. Science, 1999, 284(5413): 455-461. doi: 10.1126/science.284.5413.455
[26] Richards FM, Schofield PN, Fleming S, et al. Expression of the von Hippel-Lindau disease tumour suppressor gene during human embryogenesis[J]. Hum Mol Genet, 1996, 5(5): 639-644. doi: 10.1093/hmg/5.5.639
[27] Mitch WE, Goldberg AL. Mechanisms of muscle wasting. The role of the ubiquitin-proteasome pathway[J]. N Engl J Med, 1996, 335(25): 1897-1905. doi: 10.1056/NEJM199612193352507
[28] Hoffman MA, Ohh M, Yang H, et al. von Hippel-Lindau protein mutants linked to type 2C VHL disease preserve the ability to downregulate HIF[J]. Hum Mol Genet, 2001, 10(10): 1019-1027. doi: 10.1093/hmg/10.10.1019
[29] de Andrade KC, Frone MN, Wegman-Ostrosky T, et al. Variable population prevalence estimates of germline TP53 variants: A gnomAD-based analysis[J]. Hum Mutat, 2019, 40(1): 97-105. doi: 10.1002/humu.23673
[30] Wu SJ, Lin CT, Agathangelidis A, et al. Distinct molecular genetics of chronic lymphocytic leukemia in Taiwan: clinical and pathogenetic implications[J]. Haematologica, 2017, 102(6): 1085-1090. doi: 10.3324/haematol.2016.157552
[31] Doffe F, Carbonnier V, Tissier M, et al. Identification and functional characterization of new missense SNPs in the coding region of the TP53 gene[J]. Cell Death Differ, 2021, 28(5): 1477-1492. doi: 10.1038/s41418-020-00672-0
[32] Albers J, Rajski M, Schönenberger D, et al. Combined mutation of Vhl and Trp53 causes renal cysts and tumours in mice[J]. EMBO Mol Med, 2013, 5(6): 949-964. doi: 10.1002/emmm.201202231
[33] Zhao Z, Chen C, Lin J, et al. Synergy between von Hippel-Lindau and P53 contributes to chemosensitivity of clear cell renal cell carcinoma[J]. Mol Med Rep, 2016, 14(3): 2785-2790. doi: 10.3892/mmr.2016.5561
[34] Pacak K. Preoperative management of the pheochromocytoma patient[J]. J Clin Endocrinol Metab, 2007, 92(11): 4069-4079. doi: 10.1210/jc.2007-1720
[35] Fallah J, Brave MH, Weinstock C, et al. FDA Approval Summary: Belzutifan for von Hippel-Lindau Disease-Associated Tumors[J]. Clin Cancer Res, 2022, 28(22): 4843-4848. doi: 10.1158/1078-0432.CCR-22-1054
[36] Jonasch E, Donskov F, Iliopoulos O, et al. Belzutifan for Renal Cell Carcinoma in von Hippel-Lindau Disease[J]. N Engl J Med, 2021, 385(22): 2036-2046. doi: 10.1056/NEJMoa2103425
-
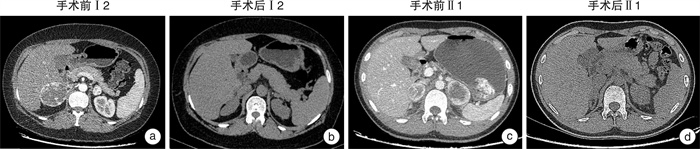
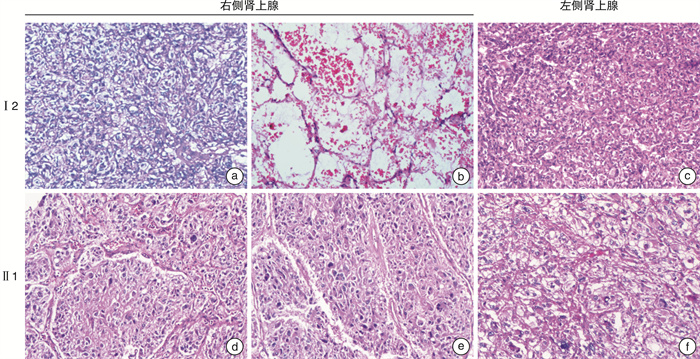
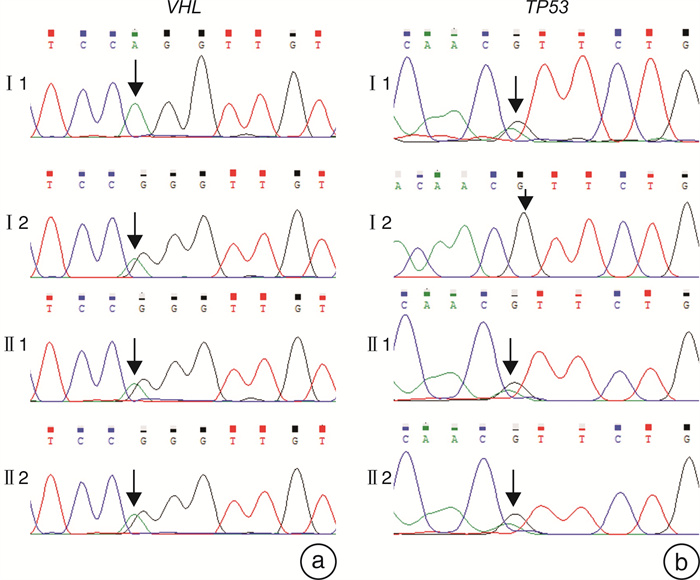
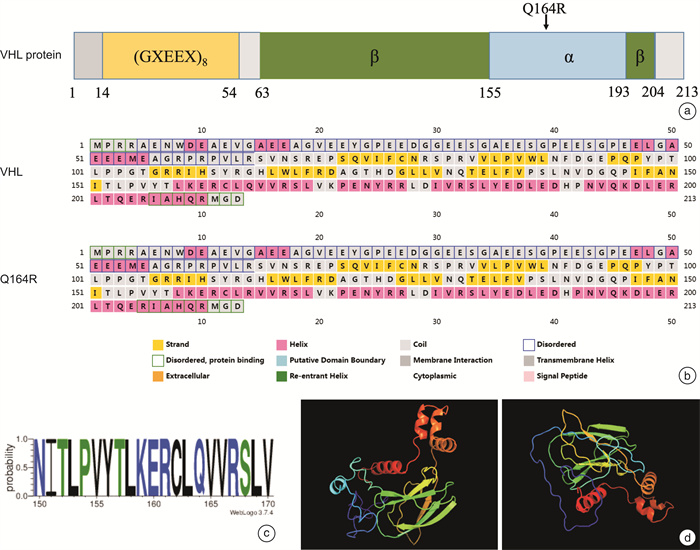
图(5)
表(1)
计量
- 文章访问数: 651
- PDF下载数: 190
- 施引文献: 0